Introducción
La enfermedad hepática esteatósica asociada a disfunción metabólica (MASLD, Metabolic dysfunction Associated Steatotic Liver Disease) se ha consolidado como la principal causa de enfermedad hepática crónica globalmente, con una prevalencia estimada en torno al 30-40% de la población adulta y una tendencia ascendente en las últimas décadas1. En 2023, un consenso multidisciplinario internacional propuso un cambio de nomenclatura de NAFLD/NASH (Non-Alcoholic Fatty Liver Disease/Non-Alcoholic Steatohepatitis) a MASLD/MASH (Metabolic dysfunction Associated Steato Hepatitis), encuadrándola bajo el paraguas de SLD (Steatotic Liver Disease). La MASLD se definió por la presencia de esteatosis (≥ 5% de hepatocitos por imagen o histología) junto con uno o más factores de riesgo cardiometabólicos y ausencia de ingesta significativa de alcohol2. Esta actualización, incorporada ya en la guía clínica de la European Association for the Study of the Liver (EASL), la European Association for the Study of Diabetes (EASD) y la European Association for the Study of Obesity (EASO) de 20243, refuerza el énfasis en la disfunción metabólica como eje patogénico y estandariza los criterios para su cribado, la estratificación de riesgo y su manejo.
La etiología de la MASLD es heterogénea y converge en la interacción de estilos de vida (dieta hipercalórica, sedentarismo), obesidad y síndrome metabólico, con contribuciones relevantes por parte de factores genéticos y epigenéticos, y de la disbiosis intestinal4–10. Además, la exposición a contaminantes ambientales se ha vinculado de manera creciente al desarrollo de esteatosis y la progresión de la enfermedad, subrayando su carácter multifactorial11–13. Por tanto, la MASLD se reconoce hoy en día como una enfermedad multisistémica que trasciende el hígado.
Desde el punto de vista fisiopatológico, la evolución de la MASLD se explica mejor mediante la hipótesis de los «múltiples impactos en paralelo», que sustituye al antiguo modelo de «dos golpes»14. En ella confluyen, de forma sincrónica, la resistencia a la insulina, la lipotoxicidad y la disfunción del eje intestino-páncreas-hígado, generando un microambiente hepático propenso a la inflamación crónica de bajo grado y la fibrosis. La resistencia a la insulina (hepática y sistémica) impulsa la lipogénesis de novo, el aumento del flujo de ácidos grasos libres al hígado y la acumulación de especies lipotóxicas (ceramidas, diacilgliceroles), con hiperinsulinemia compensatoria y crosstalk interorgánico (tejido adiposo, músculo, páncreas) que perpetúa el círculo vicioso metabólico15,16.
El estrés oxidativo y la disfunción mitocondrial constituyen nodos centrales de progresión: el exceso de sustratos y la sobrecarga lipídica alteran la función mitocondrial, favorecen la peroxidación lipídica, el estrés del retículo endoplásmico y la generación de especies reactivas de oxígeno (ROS), con daño hepatocelular e incremento de las vías de señalización profibrótica17–19. Tales procesos se amplifican por señales de patrones moleculares de daño (DAMP, Damage-Associated Molecular Patterns) y de patógenos (PAMP, Pathogen-Associated Molecular Patterns), y por metabolitos derivados de la microbiota intestinal (lipopolisacáridos, etanol endógeno y ácidos grasos de cadena corta), los cuales, al atravesar una barrera intestinal más permeable, activan vías innatas y adaptativas en el hígado10,20,21. Todo ello contribuye a la transición hacia esteatohepatitis (MASH), definida histológicamente por esteatosis con balonización, inflamación lobulillar y fibrosis (Fig. 1).
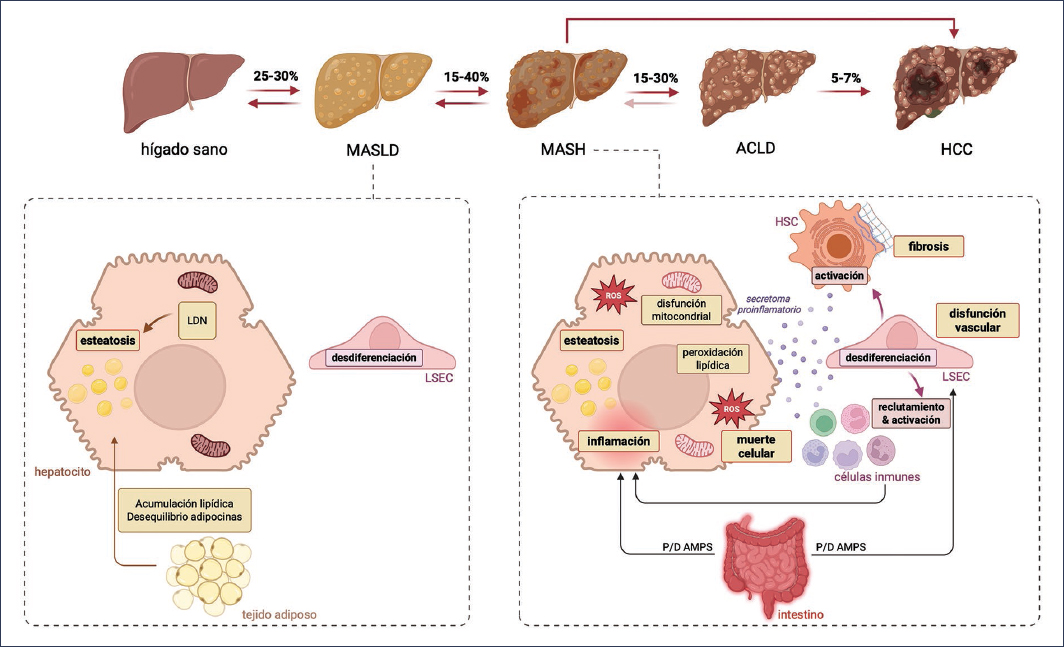
Figura 1. Fisiopatología de la enfermedad hepática asociada a disfunción metabólica. Esta figura muestra los complejos mecanismos que subyacen a la progresión desde la enfermedad hepática asociada a disfunción metabólica (MASLD) hasta la esteatohepatitis asociada a disfunción metabólica (MASH), y más allá. Entre los principales factores contribuyentes se encuentran la dislipidemia, la disfunción endotelial y la inflamación crónica, que conducen a la acumulación de lípidos en los hepatocitos, el estrés oxidativo y la disfunción mitocondrial. La figura destaca el papel de la desregulación del eje intestino-hígado y la inflamación del tejido adiposo en la progresión de la enfermedad. Además, la fibrogénesis–impulsada por la activación de las células estrelladas hepáticas y el depósito de matriz extracelular–se muestra como un sello distintivo de la transición hacia una enfermedad hepática más avanzada. Este proceso multifactorial culmina en la lesión hepática, la inflamación y la fibrosis características de la MASH. ACLD: enfermedad hepática crónica avanzada; LDN,: lipogénesis de novo; LSEC: célula endotelial sinusoidal hepática; HSC: célula estrellada hepática; P/D AMPS: patrones moleculares asociados a patógenos/peligro; ROS,: especies reactivas de oxígeno.
La persistencia de la agresión metabólicoinflamatoria precipita la muerte de los hepatocitos (a través de diversas vías, incluyendo apoptosis, necrosis y ferroptosis), la disfunción de las células endoteliales sinusoidales y el reclutamiento del compartimento inmunitario (macrófagos/células de Kupffer, monocitos, linfocitos), generando un microambiente proinflamatorio, marcado por la liberación de citocinas proinflamatorias (factor de necrosis tumoral alfa [TNFα], interleucina [IL] 1β, factor de crecimiento transformante beta [TGFβ], entre otras) que activarán las células hepáticas estrelladas22,23. Estas células sufren una transición fenotípica miofibroblástica, con propiedades proliferativas, contráctiles y migratorias, y secretan matriz extracelular en exceso, impulsando la fibrogénesis24,25. En la era de la «ómicas», a nivel de célula única, se han descrito subpoblaciones de células hepáticas estrelladas activadas con firmas fibrogénicas y proinflamatorias distintas, lo que revela la heterogeneidad funcional de este tipo celular y puede facilitar el potencial desarrollo de dianas terapéuticas específicas26,27.
Clínicamente, el grado de fibrosis es el principal predictor de eventos hepáticos y extrahepáticos y de mortalidad, por encima de otras características histológicas, lo que justificaría estrategias de estratificación no invasiva de la fibrosis y la priorización de objetivos antifibróticos28–30. La fibrosis puede progresar a cirrosis, con distorsión arquitectural y nódulos de regeneración que comprometerían la función hepática, y en una fracción de los pacientes puede progresar a carcinoma hepatocelular (HCC). Cabe señalar que el continuo MASLD-MASH-cirrosis-HCC no siempre es lineal: una proporción no desdeñable de HCC en la MASLD ocurre en pacientes sin cirrosis establecida, aunque con fibrosis avanzada en muchos casos, lo que complicaría el cribado y la detección precoz31.
Más allá del hígado, la MASLD aumenta de forma significativa el riesgo cardiovascular, siendo la principal causa de muerte en estos pacientes, y se asocia a mayor mortalidad por todas las causas, así como a un incremento de neoplasias extrahepáticas y comorbilidad cardiometabólica32,33. Este perfil de riesgo sistémico refuerza la necesidad de estrategias de manejo integradas con otras especialidades clínicas, centradas en el cambio en el estilo de vida, el control de los factores de riesgo y, cuando corresponda, terapias específicas, en línea con las guías vigentes.
En conjunto, la MASLD es un trastorno complejo y multifactorial en el que convergen lipogénesis y metabolismo lipídico desregulado, resistencia a la insulina, estrés oxidativo y del retículo endoplásmico, inflamación inmunomediada, susceptibilidad genética/epigenética y disbiosis intestinal. En las secciones siguientes de esta revisión se abordarán en profundidad los principales ejes mecanísticos con el objetivo de integrar la evidencia básica, traslacional y clínica que sustenta la fisiopatología y la progresión de la enfermedad.
Aspectos fisiopatológicos más relevantes de la MASLD
Exceso de energía y lipogénesis de novo
El exceso de energía, especialmente derivado del consumo de dietas hipercalóricas ricas en azúcares simples (como la fructosa) y grasas saturadas, constituye uno de los principales factores desencadenantes de la MASLD. Cuando la ingesta energética supera las necesidades metabólicas del organismo, el hígado actúa como un órgano de almacenamiento, acumulando triglicéridos en los hepatocitos. Este proceso se ve exacerbado por la activación de la lipogénesis de novo, una vía metabólica que convierte los hidratos de carbono en ácidos grasos.
La lipogénesis de novo está regulada por factores de transcripción, tales como SREBP-1c (Sterol Regulatory Element-Binding Protein 1c) y ChREBP (Carbohydrate Response Element-Binding Protein), que se activan en respuesta a la presencia de niveles elevados de insulina y glucosa. Estos factores inducen la expresión de enzimas clave, como la acetil coenzima A carboxilasa y la ácido graso sintasa, encargadas de la síntesis de ácidos grasos. Estos ácidos grasos se esterifican posteriormente con glicerol para formar triglicéridos, que se acumulan en el citoplasma de los hepatocitos34–36.
Además, el exceso de energía inhibe la oxidación de ácidos grasos y la secreción de lipoproteínas ricas en triglicéridos (VLDL, Very Low Density Lipoproteins), lo que contribuye aún más a la acumulación lipídica intrahepática37,38. Esta sobrecarga lipídica no solo genera esteatosis hepática, sino que además induce lipotoxicidad, un fenómeno en el que ciertos lípidos, como las ceramidas y los diacilglicéridos, alteran la función celular y generan estrés oxidativo, inflamación y apoptosis39. Por ello, este desequilibrio entre la ingesta y el gasto energético y la capacidad del hígado para manejar los lípidos representa el primer paso en la cascada patogénica de la MASLD, preparando el terreno para la progresión hacia formas más graves, como MASH y fibrosis hepática.
Resistencia a la insulina
La resistencia a la insulina es uno de los pilares fisiopatológicos más relevantes en el desarrollo de MASLD. En condiciones normales, la insulina regula el metabolismo energético promoviendo la captación de glucosa en los tejidos periféricos, inhibiendo la lipólisis en el tejido adiposo y favoreciendo la síntesis de glucógeno en el hígado. Sin embargo, en situaciones de resistencia a la insulina, frecuentemente asociadas a obesidad visceral, sedentarismo y dieta hipercalórica, estas funciones se ven comprometidas.
En el tejido adiposo, la resistencia a la insulina provoca una activación descontrolada de la lipólisis, lo que genera un aumento en la liberación de ácidos grasos libres hacia la circulación portal40. Estos ácidos grasos libres son captados por el hígado, donde se reesterifican en triglicéridos, contribuyendo a la esteatosis hepática. Además, los ácidos grasos libres pueden activar vías de señalización proinflamatorias, tales como la del factor nuclear kappa B y la JNK (Jun-N-ternimal Kinase), que interfieren con la vía de señalización de la insulina, perpetuando el estado de resistencia41,42.
En el hígado, la resistencia a la insulina altera el equilibrio entre la gluconeogénesis y la lipogénesis43,44. Aunque la insulina pierde su capacidad de inhibir la producción de glucosa hepática, mantiene su efecto lipogénico, lo que resulta en una paradoja metabólica, ya que aumenta la producción de glucosa a la vez que se acumulan lípidos. Esta situación favorece la disfunción metabólica y el desarrollo de lipotoxicidad.
A nivel molecular se ha descrito una disminución en la fosforilación del receptor de insulina y de proteínas clave como IRS-1 y Akt, lo que compromete la transducción de señales45–47. Además, la inflamación crónica de bajo grado, mediada por citocinas como el TNF-α y la IL-6, contribuye a la inhibición de esta48. En conjunto, la resistencia a la insulina no solo promueve la acumulación de grasa hepática, sino que también establece un entorno proinflamatorio y prooxidativo que facilita la progresión hacia MASH.
Alteraciones en el metabolismo lipídico
El metabolismo de los lípidos en el hígado es un proceso dinámico que involucra la captación, la síntesis, la oxidación y la exportación de ácidos grasos. En el contexto de la MASLD, este equilibrio se ve profundamente alterado, favoreciendo la acumulación de lípidos en los hepatocitos y contribuyendo al inicio y la progresión de la enfermedad.
En condiciones fisiológicas, el hígado capta ácidos grasos provenientes de la dieta, de la lipólisis del tejido adiposo y de la lipogénesis de novo49. Estos ácidos grasos pueden ser oxidados en las mitocondrias, los peroxisomas o el retículo endoplasmático, o bien ser esterificados para formar triglicéridos que se almacenan o se exportan como VLDL50.
En la MASLD, este proceso se desequilibra por varios mecanismos. En primer lugar, la captación de ácidos grasos se incrementa debido a la resistencia a la insulina y al aumento de la lipólisis periférica, lo que eleva la carga lipídica en el hígado49. Además, en segundo lugar, la oxidación de ácidos grasos se ve comprometida por la disfunción mitocondrial, lo que reduce la capacidad para eliminar el exceso de lípidos por parte del hepatocito51. Por último, la exportación de triglicéridos como VLDL se vuelve insuficiente, ya sea por defectos en la síntesis de apolipoproteínas o por sobrecarga del sistema de secreción52,53.
En conjunto, este desequilibrio favorece la acumulación de lípidos tóxicos, como ceramidas, diacilglicéridos y colesterol libre, induciendo lipotoxicidad. Estos lípidos alteran diversas vías de señalización celular, inducen estrés oxidativo e inflamación, y promueven la apoptosis de los hepatocitos. Además, la acumulación de colesterol libre en las membranas celulares puede desestabilizar los orgánulos, especialmente las mitocondrias y el retículo endoplasmático. Por tanto, se considera que la alteración del metabolismo lipídico no solo contribuye a la esteatosis hepática, sino que también actúa como un motor de progresión hacia fases avanzadas de la enfermedad.
Estrés oxidativo y disfunción mitocondrial
Como ya se ha comentado, el estrés oxidativo y la disfunción mitocondrial son elementos clave en la progresión de la MASLD desde la esteatosis simple a la MASH y la fibrosis hepática. La acumulación excesiva de lípidos en los hepatocitos, en especial de ácidos grasos libres y colesterol, sobrecarga las mitocondrias, que son las encargadas de la β-oxidación de los ácidos grasos. Esta sobrecarga genera un aumento en la producción de ROS, superando la capacidad antioxidante celular54–57.
Las ROS, tales como el peróxido de hidrógeno (H2O2), el anión superóxido (O2−) y los radicales hidroxilo (OH·), pueden dañar a los lípidos, las proteínas y los ácidos nucleicos, alterando la integridad de las membranas celulares y organelares58. En el hígado, este daño se traduce en peroxidación lipídica, disfunción del retículo endoplasmático, activación de vías inflamatorias y muerte celular por apoptosis o necrosis59.
La disfunción mitocondrial también implica una alteración en la dinámica mitocondrial (fusión y fisión), pérdida del potencial de membrana, disminución de la fosforilación oxidativa y liberación de citocromo c, lo que activa la cascada apoptótica. Además, las mitocondrias dañadas liberan señales de peligro, como los DAMP, que activan el sistema inmunitario innato, incluyendo el inflamasoma NLRP360.
Otro aspecto relevante es la disminución de la actividad de enzimas antioxidantes, como la superóxido dismutasa, la catalasa y la glutatión peroxidasa, lo que agrava el desequilibrio redox61. El estrés oxidativo también puede interferir con la función de receptores nucleares, entre ellos PPAR-α (Peroxisome Proliferator-Activated Receptor Alpha) y FXR (Farnesoid X Receptor), que regulan el metabolismo lipídico y la inflamación62.
Por tanto, el estrés oxidativo y la disfunción mitocondrial no solo contribuyen al daño hepatocelular directo, sino que también amplifican la respuesta inflamatoria y fibrogénica, y son elementos centrales en la transición de MASLD hacia estadios más graves.
Inflamación y activación del inflamasoma
La inflamación es un componente central en la progresión de la MASLD, ya que el hígado se convierte en un órgano inmunitariamente activo, donde múltiples señales de daño celular y metabólico desencadenan respuestas inflamatorias tanto innatas como adaptativas. Uno de los mecanismos más relevantes es la activación del inflamasoma, en especial el complejo NLRP3, que actúa como sensor de estrés celular63–65.
El inflamasoma NLRP3 se activa en respuesta a señales de peligro (DAMP y PAMP), como endotoxinas bacterianas, cristales de colesterol, ácidos grasos saturados y trifosfato de adenosina extracelular. Su activación conduce a la activación de la caspasa-1, que a su vez procesa las proformas de la IL-1β y la IL-18 en sus formas activas. Estas citocinas son potentes mediadores inflamatorios que amplifican la necroinflamación hepática, favoreciendo el reclutamiento de células inmunitarias como neutrófilos, monocitos y linfocitos63,64,66,67.
Además, esta inflamación en la MASLD está modulada por la polarización de los macrófagos hepáticos (también conocidos como células de Kupffer) hacia un fenotipo proinflamatorio (M1), que secreta TNF-α, IL-6 y otras citocinas que interfieren con la señalización de la insulina y promueven el daño hepatocelular68,69. Esta inflamación crónica de bajo grado también afecta a las células endoteliales sinusoidales y a las células hepáticas estrelladas, facilitando su activación y contribuyendo a la fibrogénesis70.
La activación del inflamasoma, además de tener implicaciones locales en el hígado, también puede generar efectos sistémicos, como resistencia a la insulina, disfunción endotelial y alteraciones en otros órganos71,72, por lo que la combinación de inflamación y activación del inflamasoma representa un puente entre el metabolismo alterado y la respuesta inmunitaria, siendo mecanismos fundamentales en la progresión de la MASLD.
Factores genéticos
Los factores genéticos desempeñan un papel fundamental en la susceptibilidad individual al desarrollo y la progresión de enfermedad73. Aunque los factores ambientales y metabólicos son determinantes clave, la variabilidad genética explica por qué algunos individuos desarrollan formas graves de enfermedad incluso en ausencia de factores de riesgo metabólico, mientras que otros presentan esteatosis leve sin progresión de la enfermedad pese a tener múltiples factores de riesgo.
Entre los genes más estudiados se encuentra PNPLA3 (Patatin-Like Phospholipase Domain-Containing Protein 3), cuyo polimorfismo rs738409 (I148M) se asocia fuertemente con acumulación de grasa hepática, inflamación y fibrosis, así como con un riesgo incrementado de desarrollar cirrosis y HCC. Este alelo mutado reduce la actividad lipasa de la proteína, favoreciendo la retención de triglicéridos en los hepatocitos. Su efecto es independiente de la resistencia a la insulina, lo que le convierte en un robusto marcador genético de riesgo74,75.
Otro gen relevante es TM6SF2 (Transmembrane 6 Superfamily Member 2), cuyo polimorfismo rs 58542926 (E167K) se asocia con una disminución en la secreción de VLDL, lo que favorece la acumulación de lípidos intrahepáticos76. Sin embargo, aunque este gen parece proteger contra enfermedades cardiovasculares al reducir los niveles plasmáticos de lípidos, se ha demostrado que aumenta el riesgo de fibrosis hepática77.
El gen MBOAT7 (Membrane-Bound O-Acyltransferase Domain-Containing 7) también ha sido implicado en la regulación del metabolismo de fosfolípidos. Su variante rs641738 se asocia con inflamación y fibrosis, posiblemente por alterar la composición de las membranas celulares y la señalización intracelular78.
Más recientemente, se ha identificado el gen HSD17B13, variante rs72613567, cuya pérdida de función parece tener un efecto protector en la progresión hacia MASH y fibrosis79–81. Este hallazgo ha abierto nuevas vías terapéuticas basadas en la modulación genética.
Los factores genéticos no solo modulan la expresión clínica y fenotípica de la MASLD, sino que ofrecen oportunidades para la medicina personalizada, permitiendo identificar individuos de alto riesgo y desarrollar estrategias terapéuticas dirigidas.
Disbiosis y permeabilidad intestinal
El eje intestino-hígado desempeña un papel crucial en la fisiopatología de las enfermedades hepáticas, en concreto de la MASLD, y su alteración por disbiosis intestinal y aumento de la permeabilidad de la barrera intestinal contribuye significativamente a la progresión82. El intestino alberga una comunidad microbiana diversa que participa en la digestión, el metabolismo de nutrientes, la modulación inmunitaria y la protección de la barrera epitelial83. En la MASLD, esta microbiota se ve alterada tanto en su composición como en su función, fenómeno conocido como disbiosis.
La disbiosis intestinal se caracteriza por una disminución de bacterias beneficiosas (como Faecalibacterium prausnitzii y Akkermansia muciniphila) y un aumento de especies proinflamatorias (como Enterobacteriaceae y Proteobacteria)84,85. Esta alteración favorece la producción de metabolitos nocivos, como etanol endógeno, y el desequilibrio de la proporción de ácidos grasos de cadena corta, amonio, y lipopolisacáridos, que pueden atravesar la barrera intestinal cuando esta se encuentra comprometida, fenómeno conocido como intestino permeable (leaky gut)86.
La permeabilidad intestinal aumentada permite el paso de endotoxinas y otros productos bacterianos hacia la circulación portal, alcanzando el hígado y activando receptores como los TLR4 (Toll-like Receptor 4) en las células de Kupffer y los hepatocitos. Esta activación desencadena respuestas inflamatorias, producción de citocinas y activación del inflamasoma, contribuyendo a la necroinflamación hepática y a la progresión hacia MASH87.
Además, la disbiosis puede influir en el metabolismo de los ácidos biliares, alterando la señalización de receptores como FXR y TGR5 (Takeda G Protein-Coupled Receptor 5), que regulan la homeostasis energética, la inflamación y la sensibilidad a la insulina88. También se ha observado que ciertos perfiles microbianos pueden modular la expresión de genes hepáticos relacionados con la lipogénesis y la fibrosis, poniendo de relevancia el papel crucial de la disbiosis intestinal en la progresión de la enfermedad89.
Disfunción microcirculatoria hepática y fibrosis
La disfunción microcirculatoria hepática representa una consecuencia directa de la interacción de los mecanismos fisiopatológicos descritos en la MASLD. El hígado posee una arquitectura vascular única, compuesta por sinusoides que permiten el intercambio eficiente de nutrientes, metabolitos y células inmunitarias entre la sangre portal y los hepatocitos. En la MASLD, esta microcirculación se ve progresivamente alterada, contribuyendo al deterioro funcional del órgano y al desarrollo de fibrosis.
La acumulación de lípidos en los hepatocitos, el estrés oxidativo y la inflamación crónica inducen daño endotelial en las células endoteliales sinusoidales. Estas células pierden su fenotipo especializado, caracterizado por fenestraciones y baja expresión de moléculas de adhesión, y adoptan un estado proinflamatorio, vasoconstrictor y procoagulante90. Esta transformación aumenta el tono vascular hepático y reduce el intercambio entre la sangre y los hepatocitos, promoviendo hipoxia local y disfunción metabólica. A su vez, la hipoxia activa factores, como HIF-1α (Hypoxia-Inducible Factor 1-alpha), que promueven la angiogénesis patológica y la activación de células hepáticas estrelladas. Estas células, en respuesta a citocinas inflamatorias (TGF-β, factor de crecimiento derivado de plaquetas, IL-1β), se diferencian en miofibroblastos, adquiriendo propiedades contráctiles, migratorias y secretoras. El resultado es una producción excesiva de componentes de la matriz extracelular (colágeno tipo I y III, fibronectina), que se depositan en el espacio de Disse, alterando aún más la microcirculación91,92.
La fibrosis hepática se caracteriza por un desequilibrio entre la síntesis y la degradación de la matriz extracelular, debido a la inhibición de metaloproteinasas y la activación de sus inhibidores93. Este proceso distorsiona la arquitectura hepática, compromete el flujo sanguíneo portal y puede evolucionar hacia cirrosis, con formación de nódulos regenerativos y pérdida de función hepática. La fibrosis, sumada al aumento en el tono vascular derivado de la desdiferenciación de las células endoteliales sinusoidales y la activación de las células hepáticas estrelladas, conlleva un aumento de la resistencia vascular intrahepática, factor primario en el derivado desarrollo de hipertensión portal94.
Conclusiones
La MASLD representa un desafío clínico y científico de creciente relevancia, dada su alta prevalencia global y su carácter multisistémico. A lo largo de esta revisión se ha analizado su carácter dinámico y multifactorial, siendo el resultado de una compleja interacción de factores genéticos, ambientales, metabólicos y celulares que convergen en una cascada de eventos patológicos.
Además, la progresión fisiopatológica de la MASLD desde la esteatosis hepática hasta la fibrosis avanzada tiene implicaciones clínicas significativas que afectan no solo al hígado, sino también al estado general del paciente, cuyas consecuencias clínicas derivan de la interacción de alteraciones metabólicas, inflamatorias, genéticas, inmunitarias y microcirculatorias95.
Sistémicamente se asocia con un aumento significativo del riesgo de enfermedad cardiovascular, que representa la principal causa de muerte en estos pacientes. La resistencia a la insulina, la dislipidemia, la inflamación crónica y la disfunción endotelial contribuyen al desarrollo de aterosclerosis, hipertensión y eventos cardiovasculares mayores96–99. Además, se ha observado una mayor incidencia de diabetes mellitus tipo 2, síndrome metabólico y algunos tipos de cáncer extrahepáticos, como el colorrectal y el de mama100–103. La disbiosis intestinal y la inflamación sistémica también pueden afectar la función renal, el sistema nervioso central y el estado inmunitario general82,104,105.
En el hígado, la acumulación de grasa y el daño celular progresivo pueden evolucionar hacia MASH, caracterizada por necroinflamación, balonización hepatocelular y activación de las células hepáticas estrelladas25,106. Esta etapa puede conducir hacia el desarrollo de fibrosis, que puede avanzar hacia cirrosis hepática, con distorsión de la arquitectura hepática, hipertensión portal, insuficiencia hepática y riesgo de HCC92,107. Es importante destacar que esta progresión no siempre es lineal, y algunos pacientes pueden desarrollar HCC en ausencia de cirrosis, es decir, en etapas tempranas de una enfermedad hepática crónica avanzada.
Además, esta patología se asocia con un aumento significativo del riesgo cardiovascular, metabólico y neoplásico, lo que refuerza la necesidad de abordajes diagnósticos y terapéuticos integrales. La identificación de biomarcadores específicos, el estudio de variantes genéticas y el análisis del microbioma intestinal abren nuevas vías para la medicina personalizada y la prevención de complicaciones. En este escenario complejo, la medicina personalizada emerge como una estrategia prometedora para mejorar el manejo clínico de la enfermedad. La identificación de variantes genéticas asociadas a mayor susceptibilidad, junto con el análisis fenotípico del paciente, permite una estratificación más precisa del riesgo y una selección más racional de las intervenciones terapéuticas. Además, el desarrollo de biomarcadores no invasivos y de herramientas de diagnóstico molecular facilitará la monitorización dinámica de la enfermedad y la evaluación de la respuesta al tratamiento. En conjunto, la medicina personalizada no solo optimiza la eficacia clínica, sino que también reduce el riesgo de efectos adversos y mejora la calidad de vida de los pacientes, posicionándose como un pilar fundamental en el futuro del tratamiento de la MASLD108.
Finalmente, desde el punto de vista clínico, estas consecuencias requieren un enfoque multidisciplinario en el manejo del paciente con MASLD, que incluirá hepatología, endocrinología, cardiología y nutrición. La identificación precoz de los pacientes en riesgo, el uso de biomarcadores no invasivos y la implementación de estrategias terapéuticas personalizadas son esenciales para prevenir las complicaciones y mejorar el pronóstico, reduciendo su impacto en la carga económica de los sistemas sanitarios públicos.
Financiamiento
Los autores agradecen la financiación continuada por parte del Instituto de Salud Carlos III (ISCIII; actualmente PI23/00945, DTS22/00010, DTS24/00035 y AC24/00124 a J. Gracia-Sancho; CD21/00095, PI22/01342 y PI24/02008 a R. Gallego-Durán, todos cofinanciados por la Unión Europea), AGAUR-Generalitat de Catalunya (2021 SGR 01322, 2021 PROD 00036, 2025 PROD 00184 y 2025 INNOV 00043) a J. Gracia-Sancho, y el CIBEREHD (financiado por el Instituto de Salud Carlos III).
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Consideraciones éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad, consentimiento informado y aprobación ética. El estudio no involucra datos personales de pacientes ni requiere aprobación ética. No se aplican las guías SAGER.
Declaración sobre el uso de inteligencia artificial. Los autores declaran que no utilizaron ningún tipo de inteligencia artificial generativa para la redacción de este manuscrito.
