Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) has consolidated as the main cause of chronic liver disease globally, with an estimated prevalence of around 30-40% of the adult population and an increasing trend in recent decades1. In 2023, an international multidisciplinary consensus proposed a nomenclature change from NAFLD/NASH (non-alcoholic fatty liver disease/non-alcoholic steatohepatitis) to MASLD/MASH (metabolic dysfunction-associated steatotic liver disease/metabolic dysfunction-associated steatohepatitis), framing it under the umbrella of SLD (steatotic liver disease). MASLD was defined by the presence of steatosis (≥ 5% of hepatocytes by imaging or histology) along with one or more cardiometabolic risk factors and absence of significant alcohol intake2. This update, already incorporated in the 2024 clinical guideline of the European Association for the Study of the Liver (EASL), the European Association for the Study of Diabetes (EASD), and the European Association for the Study of Obesity (EASO)3, reinforces the emphasis on metabolic dysfunction as the pathogenic axis and standardizes the criteria for its screening, risk stratification, and management.
The etiology of MASLD is heterogeneous and converges in the interaction of lifestyle factors (hypercaloric diet, sedentary lifestyle), obesity, and metabolic syndrome, with relevant contributions from genetic and epigenetic factors, and intestinal dysbiosis4–10. In addition, exposure to environmental pollutants has been increasingly linked to the development of steatosis and disease progression, underscoring its multifactorial nature11–13. Therefore, MASLD is recognized today as a multisystemic disease that transcends the liver.
From a pathophysiological standpoint, the evolution of MASLD is best explained by the “multiple parallel hits” hypothesis, which replaces the old “two-hit” model14. It converges, synchronously, insulin resistance, lipotoxicity, and dysfunction of the gut-pancreas-liver axis, generating a hepatic microenvironment prone to chronic low-grade inflammation and fibrosis. Insulin resistance (hepatic and systemic) drives de novo lipogenesis, increased flux of free fatty acids to the liver, and accumulation of lipotoxic species (ceramides, diacylglycerols), with compensatory hyperinsulinemia and inter-organ crosstalk (adipose tissue, muscle, pancreas) that perpetuates the metabolic vicious cycle15,16.
Oxidative stress and mitochondrial dysfunction constitute central nodes of progression: substrate excess and lipid overload alter mitochondrial function, favor lipid peroxidation, endoplasmic reticulum stress, and generation of reactive oxygen species (ROS), with hepatocellular damage and increased profibrotic signaling pathways17–19. Such processes are amplified by damage-associated molecular pattern (DAMP) and pathogen-associated molecular pattern (PAMP) signals, and by metabolites derived from the intestinal microbiota (lipopolysaccharides, endogenous ethanol, and short-chain fatty acids), which, when crossing a more permeable intestinal barrier, activate innate and adaptive pathways in the liver10,20,21. All this contributes to the transition toward steatohepatitis (MASH), defined histologically by steatosis with ballooning, lobular inflammation, and fibrosis (Fig. 1).
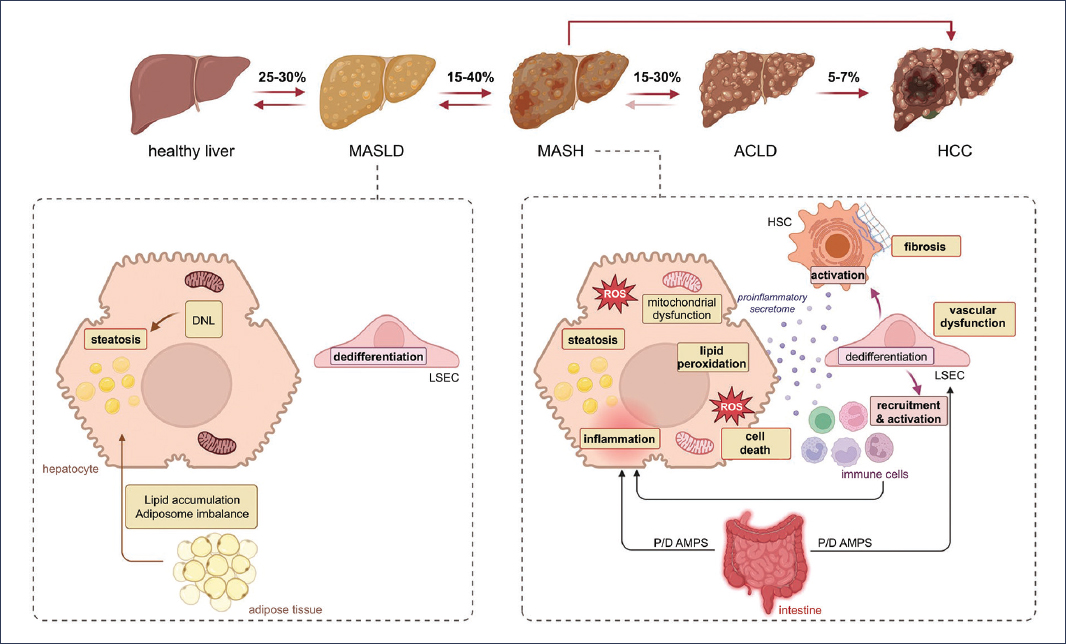
Figure 1. Pathophysiology of liver disease associated with metabolic dysfunction. This figure shows the complex mechanisms that lead to progression from metabolic dysfunction-associated steatotic liver disease (MASLD) to metabolic dysfunction-associated steatohepatitis (MASH), and beyond. Among the main contributing factors are dyslipidemia, endothelial dysfunction, and chronic inflammation, which lead to the accumulation of lipids in the hepatocyte. In more advanced stages, mitochondrial dysfunction plays a key role in the progression of the disease. Additionally, the figure highlights the role of dysregulation of the intestine-liver axis and inflammation of adipose tissue in disease progression. Furthermore, fibrogenesis – driven by the activation of hepatic stellate cells and the deposition of extracellular matrix – is shown as a distinctive hallmark of the transition to more advanced liver disease. This multifactorial process culminates in liver injury, inflammation, and fibrosis characteristic of MASH. ACLD: advanced chronic liver disease; DNL: de novo lipogenesis; LSEC: liver sinusoidal endothelial cell; HSC: hepatic stellate cell; P/D AMPS: pathogen/danger-associated molecular patterns; ROS: reactive oxygen species.
The persistence of metabolic-inflammatory injury precipitates hepatocyte death (through various pathways, including apoptosis, necrosis, and ferroptosis), sinusoidal endothelial cell dysfunction, and recruitment of the immune compartment (macrophages/Kupffer cells, monocytes, lymphocytes), generating a proinflammatory microenvironment, marked by the release of proinflammatory cytokines (tumor necrosis factor alpha [TNFα], interleukin [IL] 1β, transforming growth factor beta [TGFβ], among others) that will activate hepatic stellate cells22,23. These cells undergo a myofibroblastic phenotypic transition, with proliferative, contractile, and migratory properties, and secrete excessive extracellular matrix, driving fibrogenesis24,25. In the “omics” era, at the single-cell level, subpopulations of activated hepatic stellate cells with distinct fibrogenic and proinflammatory signatures have been described, revealing the functional heterogeneity of this cell type and potentially facilitating the development of specific therapeutic targets26,27.
Clinically, the degree of fibrosis is the main predictor of hepatic and extrahepatic events and mortality, above other histological features, which would justify non-invasive fibrosis stratification strategies and prioritization of antifibrotic objectives28–30. Fibrosis can progress to cirrhosis, with architectural distortion and regenerative nodules that would compromise hepatic function, and in a fraction of patients, can progress to hepatocellular carcinoma (HCC). It should be noted that the MASLD-MASH-cirrhosis-HCC continuum is not always linear: a non-negligible proportion of HCC in MASLD occurs in patients without established cirrhosis, although with advanced fibrosis in many cases, which would complicate screening and early detection31.
Beyond the liver, MASLD significantly increases cardiovascular risk, being the main cause of death in these patients, and is associated with higher all-cause mortality, as well as an increase in extrahepatic neoplasms and cardiometabolic comorbidity32,33. This systemic risk profile reinforces the need for integrated management strategies with other clinical specialties, focused on lifestyle change, control of risk factors, and, when appropriate, specific therapies, in line with current guidelines.
Overall, MASLD is a complex and multifactorial disorder in which dysregulated lipogenesis and lipid metabolism, insulin resistance, oxidative and endoplasmic reticulum stress, immune-mediated inflammation, genetic/epigenetic susceptibility, and intestinal dysbiosis converge. The following sections of this review will address in depth the main mechanistic axes with the objective of integrating basic, translational, and clinical evidence that supports the pathophysiology and progression of the disease.
Most relevant pathophysiological aspects of MASLD
Energy excess and de novo lipogenesis
Energy excess, especially derived from the consumption of hypercaloric diets rich in simple sugars (such as fructose) and saturated fats, constitutes one of the main triggering factors of MASLD. When energy intake exceeds the metabolic needs of the organism, the liver acts as a storage organ, accumulating triglycerides in hepatocytes. This process is exacerbated by the activation of de novo lipogenesis, a metabolic pathway that converts carbohydrates into fatty acids.
De novo lipogenesis is regulated by transcription factors, such as SREBP-1c (sterol regulatory element-binding protein 1c) and ChREBP (carbohydrate response element-binding protein), which are activated in response to the presence of elevated levels of insulin and glucose. These factors induce the expression of key enzymes, such as acetyl coenzyme A carboxylase and fatty acid synthase, responsible for fatty acid synthesis. These fatty acids are subsequently esterified with glycerol to form triglycerides, which accumulate in the cytoplasm of hepatocytes34–36.
In addition, energy excess inhibits fatty acid oxidation and the secretion of triglyceride-rich lipoproteins (VLDL, very low density lipoproteins), further contributing to intrahepatic lipid accumulation37,38. This lipid overload not only generates hepatic steatosis but also induces lipotoxicity, a phenomenon in which certain lipids, such as ceramides and diacylglycerols, alter cellular function and generate oxidative stress, inflammation, and apoptosis39. Therefore, this imbalance between intake and energy expenditure and the liver’s capacity to handle lipids represents the first step in the pathogenic cascade of MASLD, setting the stage for progression toward more severe forms, such as MASH and hepatic fibrosis.
Insulin resistance
Insulin resistance is one of the most relevant pathophysiological pillars in the development of MASLD. Under normal conditions, insulin regulates energy metabolism by promoting glucose uptake in peripheral tissues, inhibiting lipolysis in adipose tissue, and favoring glycogen synthesis in the liver. However, in situations of insulin resistance, frequently associated with visceral obesity, sedentary lifestyle, and hypercaloric diet, these functions are compromised.
In adipose tissue, insulin resistance causes uncontrolled activation of lipolysis, which generates an increase in the release of free fatty acids into the portal circulation40. These free fatty acids are taken up by the liver, where they are re-esterified into triglycerides, contributing to hepatic steatosis. In addition, free fatty acids can activate proinflammatory signaling pathways, such as that of nuclear factor kappa B and JNK (Jun-N-terminal kinase), which interfere with the insulin signaling pathway, perpetuating the state of resistance41,42.
In the liver, insulin resistance alters the balance between gluconeogenesis and lipogenesis43,44. Although insulin loses its capacity to inhibit hepatic glucose production, it maintains its lipogenic effect, resulting in a metabolic paradox, since glucose production increases while lipids accumulate. This situation favors metabolic dysfunction and the development of lipotoxicity.
At the molecular level, a decrease in the phosphorylation of the insulin receptor and key proteins such as IRS-1 and Akt has been described, which compromises signal transduction45–47. In addition, chronic low-grade inflammation, mediated by cytokines such as TNF-α and IL-6, contributes to its inhibition48. Overall, insulin resistance not only promotes hepatic fat accumulation but also establishes a proinflammatory and prooxidative environment that facilitates progression toward MASH.
Lipid metabolism disturbances
Lipid metabolism in the liver is a dynamic process that involves the uptake, synthesis, oxidation, and export of fatty acids. In the context of MASLD, this balance is profoundly altered, favoring lipid accumulation in hepatocytes and contributing to the onset and progression of the disease.
Under physiological conditions, the liver takes up fatty acids from the diet, from adipose tissue lipolysis, and from de novo lipogenesis49. These fatty acids can be oxidized in the mitochondria, peroxisomes, or endoplasmic reticulum, or be esterified to form triglycerides that are stored or exported as VLDL50.
In MASLD, this process becomes unbalanced by several mechanisms. First, fatty acid uptake increases due to insulin resistance and increased peripheral lipolysis, which elevates the lipid load in the liver49. Second, fatty acid oxidation is compromised by mitochondrial dysfunction, which reduces the capacity to eliminate excess lipids by the hepatocyte51. Finally, triglyceride export as VLDL becomes insufficient, either due to defects in apolipoprotein synthesis or secretion system overload52,53.
Overall, this imbalance favors the accumulation of toxic lipids, such as ceramides, diacylglycerols, and free cholesterol, inducing lipotoxicity. These lipids alter various cellular signaling pathways, induce oxidative stress and inflammation, and promote hepatocyte apoptosis. In addition, the accumulation of free cholesterol in cell membranes can destabilize organelles, especially mitochondria and the endoplasmic reticulum. Therefore, the alteration of lipid metabolism not only contributes to hepatic steatosis but also acts as a driver of progression toward advanced stages of the disease.
Oxidative stress and mitochondrial dysfunction
As previously mentioned, oxidative stress and mitochondrial dysfunction are key elements in the progression of MASLD from simple steatosis to MASH and hepatic fibrosis. Excessive accumulation of lipids in hepatocytes, especially free fatty acids and cholesterol, overloads the mitochondria, which are responsible for the β-oxidation of fatty acids. This overload generates an increase in ROS production, exceeding cellular antioxidant capacity54–57.
ROS, such as hydrogen peroxide (H2O2), superoxide anion (O2–), and hydroxyl radicals (OH·), can damage lipids, proteins, and nucleic acids, altering the integrity of cellular and organellar membranes58. In the liver, this damage translates into lipid peroxidation, endoplasmic reticulum dysfunction, activation of inflammatory pathways, and cell death by apoptosis or necrosis59.
Mitochondrial dysfunction also involves an alteration in mitochondrial dynamics (fusion and fission), loss of membrane potential, decreased oxidative phosphorylation, and release of cytochrome c, which activates the apoptotic cascade. In addition, damaged mitochondria release danger signals, such as DAMPs, which activate the innate immune system, including the NLRP3 inflammasome60.
Another relevant aspect is the decreased activity of antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, which aggravates the redox imbalance61. Oxidative stress can also interfere with the function of nuclear receptors, among them PPAR-α (peroxisome proliferator-activated receptor alpha) and FXR (farnesoid x receptor), which regulate lipid metabolism and inflammation62.
Therefore, oxidative stress and mitochondrial dysfunction not only contribute to direct hepatocellular damage, but also amplify the inflammatory and fibrogenic response, and are central elements in the transition from MASLD toward more severe stages.
Inflammation and inflammasome activation
Inflammation is a central component in the progression of MASLD, as the liver becomes an immunologically active organ, where multiple signals of cellular and metabolic damage trigger both innate and adaptive inflammatory responses. One of the most relevant mechanisms is inflammasome activation, especially the NLRP3 complex, which acts as a sensor of cellular stress63–65.
The NLRP3 inflammasome is activated in response to danger signals (DAMPs and PAMPs), such as bacterial endotoxins, cholesterol crystals, saturated fatty acids, and extracellular adenosine triphosphate. Its activation leads to caspase-1 activation, which in turn processes the proforms of IL-1β and IL-18 into their active forms. These cytokines are potent inflammatory mediators that amplify hepatic necroinflammation, favoring the recruitment of immune cells such as neutrophils, monocytes, and lymphocytes63,64,66,67.
In addition, inflammation in MASLD is modulated by the polarization of hepatic macrophages (also known as Kupffer cells) toward a proinflammatory phenotype (M1), which secretes TNF-α, IL-6, and other cytokines that interfere with insulin signaling and promote hepatocellular damage68,69. This chronic low-grade inflammation also affects sinusoidal endothelial cells and hepatic stellate cells, facilitating their activation and contributing to fibrogenesis70.
Inflammasome activation, in addition to having local implications in the liver, can also generate systemic effects, such as insulin resistance, endothelial dysfunction, and alterations in other organs71,72, so that the combination of inflammation and inflammasome activation represents a bridge between altered metabolism and the immune response, being fundamental mechanisms in the progression of MASLD.
Genetic factors
Genetic factors play a fundamental role in individual susceptibility to the development and progression of disease73. Although environmental and metabolic factors are key determinants, genetic variability explains why some individuals develop severe forms of disease even in the absence of metabolic risk factors, while others present mild steatosis without disease progression despite having multiple risk factors.
Among the most studied genes is PNPLA3 (patatin-like phospholipase domain-containing protein 3), whose polymorphism rs738409 (I148M) is strongly associated with hepatic fat accumulation, inflammation, and fibrosis, as well as an increased risk of developing cirrhosis and HCC. This mutant allele reduces the lipase activity of the protein, favoring triglyceride retention in hepatocytes. Its effect is independent of insulin resistance, making it a robust genetic risk marker74,75.
Another relevant gene is TM6SF2 (transmembrane 6 superfamily member 2), whose polymorphism rs58542926 (E167K) is associated with a decrease in VLDL secretion, which favors the accumulation of intrahepatic lipids76. However, although this gene appears to protect against cardiovascular diseases by reducing plasma lipid levels, it has been shown to increase the risk of hepatic fibrosis77.
The gene MBOAT7 (membrane-bound o-acyltransferase domain-containing 7) has also been implicated in the regulation of phospholipid metabolism. Its variant rs641738 is associated with inflammation and fibrosis, possibly by altering the composition of cell membranes and intracellular signaling78.
More recently, the gene HSD17B13, variant rs72613567, has been identified; its loss of function appears to have a protective effect in progression toward MASH and fibrosis79–81. This finding has opened new therapeutic avenues based on genetic modulation.
Genetic factors not only modulate the clinical and phenotypic expression of MASLD, but also offer opportunities for personalized medicine, allowing identification of high-risk individuals and development of targeted therapeutic strategies.
Dysbiosis and intestinal permeability
The gut-liver axis plays a crucial role in the pathophysiology of liver diseases, specifically MASLD, and its alteration by intestinal dysbiosis and increased intestinal barrier permeability contributes significantly to progression82. The intestine harbors a diverse microbial community that participates in digestion, nutrient metabolism, immune modulation, and epithelial barrier protection83. In MASLD, this microbiota is altered both in its composition and function, a phenomenon known as dysbiosis.
Intestinal dysbiosis is characterized by a decrease in beneficial bacteria (such as Faecalibacterium prausnitzii and Akkermansia muciniphila) and an increase in proinflammatory species (such as Enterobacteriaceae and Proteobacteria)84,85. This alteration favors the production of harmful metabolites, such as endogenous ethanol, and the imbalance in the proportion of short-chain fatty acids, ammonia, and lipopolysaccharides, which can cross the intestinal barrier when it is compromised, a phenomenon known as leaky gut86.
Increased intestinal permeability allows the passage of endotoxins and other bacterial products into the portal circulation, reaching the liver and activating receptors such as TLR4 (toll-like receptor 4) in Kupffer cells and hepatocytes. This activation triggers inflammatory responses, cytokine production, and inflammasome activation, contributing to hepatic necroinflammation and progression toward MASH87.
In addition, dysbiosis can influence bile acid metabolism, altering the signaling of receptors such as FXR and TGR5 (Takeda G protein-coupled receptor 5), which regulate energy homeostasis, inflammation, and insulin sensitivity88. It has also been observed that certain microbial profiles can modulate the expression of hepatic genes related to lipogenesis and fibrosis, highlighting the crucial role of intestinal dysbiosis in disease progression89.
Hepatic microcirculatory dysfunction and fibrosis
Hepatic microcirculatory dysfunction represents a direct consequence of the interaction of the pathophysiological mechanisms described in MASLD. The liver possesses a unique vascular architecture, composed of sinusoids that allow efficient exchange of nutrients, metabolites, and immune cells between portal blood and hepatocytes. In MASLD, this microcirculation is progressively altered, contributing to the functional deterioration of the organ and the development of fibrosis.
Lipid accumulation in hepatocytes, oxidative stress, and chronic inflammation induce endothelial damage in sinusoidal endothelial cells. These cells lose their specialized phenotype, characterized by fenestrations and low expression of adhesion molecules, and adopt a proinflammatory, vasoconstrictive, and procoagulant state90. This transformation increases hepatic vascular tone and reduces exchange between blood and hepatocytes, promoting local hypoxia and metabolic dysfunction. In turn, hypoxia activates factors, such as HIF-1β (hypoxia-inducible factor 1-alpha), which promote pathological angiogenesis and hepatic stellate cell activation. These cells, in response to inflammatory cytokines (TGF-β, platelet-derived growth factor, IL-1β), differentiate into myofibroblasts, acquiring contractile, migratory, and secretory properties. The result is excessive production of extracellular matrix components (collagen type I and III, fibronectin), which are deposited in the space of Disse, further altering microcirculation91,92.
Hepatic fibrosis is characterized by an imbalance between the synthesis and degradation of extracellular matrix, due to the inhibition of metalloproteinases and activation of their inhibitors93. This process distorts hepatic architecture, compromises portal blood flow, and can evolve toward cirrhosis, with regenerative nodule formation and loss of hepatic function. Fibrosis, combined with the increase in vascular tone derived from sinusoidal endothelial cell dedifferentiation and hepatic stellate cell activation, leads to an increase in intrahepatic vascular resistance, a primary factor in the development of portal hypertension94.
Conclusions
MASLD represents a clinical and scientific challenge of growing relevance, given its high global prevalence and its multisystemic character. Throughout this review, its dynamic and multifactorial character has been analyzed, being the result of a complex interaction of genetic, environmental, metabolic, and cellular factors that converge in a cascade of pathological events.
Moreover, the pathophysiological progression of MASLD from hepatic steatosis to advanced fibrosis has significant clinical implications that affect not only the liver but also the general state of the patient, whose clinical consequences derive from the interaction of metabolic, inflammatory, genetic, immunological, and microcirculatory alterations95.
Systemically, it is associated with a significantly increased risk of cardiovascular disease, which represents the leading cause of death in these patients. Insulin resistance, dyslipidemia, chronic inflammation, and endothelial dysfunction contribute to the development of atherosclerosis, hypertension, and major cardiovascular events96–99. Furthermore, a higher incidence of type 2 diabetes mellitus, metabolic syndrome, and certain types of extrahepatic cancers, such as colorectal and breast cancer, has been observed100–103. Intestinal dysbiosis and systemic inflammation may also affect renal function, the central nervous system, and overall immune status82,104,105.
In the liver, fat accumulation and progressive cellular damage can evolve toward MASH, characterized by necroinflammation, hepatocellular ballooning, and hepatic stellate cell activation25,106. This stage can lead toward the development of fibrosis, which can advance toward hepatic cirrhosis, with distortion of hepatic architecture, portal hypertension, hepatic insufficiency, and HCC risk92,107. It is important to highlight that this progression is not always linear, and some patients can develop HCC in the absence of cirrhosis, that is, in early stages of advanced chronic liver disease.
Moreover, this pathology is associated with a significant increase in cardiovascular, metabolic, and neoplastic risk, which reinforces the need for comprehensive diagnostic and therapeutic approaches. The identification of specific biomarkers, the study of genetic variants, and the analysis of the intestinal microbiome open new avenues for personalized medicine and prevention of complications. In this complex scenario, personalized medicine emerges as a promising strategy to improve the clinical management of the disease. The identification of genetic variants associated with greater susceptibility, together with the phenotypic analysis of the patient, allows a more precise risk stratification and a more rational selection of therapeutic interventions. In addition, the development of non-invasive biomarkers and molecular diagnostic tools will facilitate dynamic disease monitoring and evaluation of treatment response. Overall, personalized medicine not only optimizes clinical efficacy, but also reduces the risk of adverse effects and improves patients’ quality of life, positioning itself as a fundamental pillar in the future of MASLD treatment108.
Finally, from the clinical point of view, these consequences require a multidisciplinary approach in the management of the patient with MASLD, which will include hepatology, endocrinology, cardiology, and nutrition. Early identification of at-risk patients, the use of non-invasive biomarkers, and the implementation of personalized therapeutic strategies are essential to prevent complications and improve prognosis, reducing their impact on the economic burden of public health systems.
Funding
The authors acknowledge continued funding from the Instituto de Salud Carlos III (ISCIII; currently PI23/00945, DTS22/00010, DTS24/00035, and AC24/00124 to J. Gracia-Sancho; CD21/00095, PI22/01342, and PI24/02008 to R. Gallego-Durán, all co-financed by the European Union), AGAUR-Generalitat de Catalunya (2021 SGR 01322, 2021 PROD 00036, 2025 PROD 00184, and 2025 INNOV 00043) to J. Gracia-Sancho, and CIBEREHD (funded by the Instituto de Salud Carlos III).
Conflicts of interest
The authors declare no conflicts of interest.
Ethical considerations
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this research.
Confidentiality, informed consent, and ethical approval. The study does not involve personal patient data nor requires ethical approval. SAGER guidelines do not apply.
Declaration on the use of artificial intelligence. The authors declare that they did not use any type of generative artificial intelligence for the writing of this manuscript.
